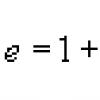Элементы биоорганической химии. Предмет биоорганической химии. классификация, строение, реакционная способность органических соединений джеймс дьюи уотсон жерар, герхардт шарль фредерик. Пабк, паск. строение. характеристика. значение
БИООРГАНИЧЕСКАЯ ХИМИЯ, изучает связь между строением органических веществ и их биологическими функциями, используя в основном методы органической и физической химии, а также физики и математики. Биоорганическая химия полностью охватывает химию природных соединений и частично пересекается с биохимией и молекулярной биологией. Объектами её изучения служат биологически важные природные соединения - главным образом биополимеры (белки, нуклеиновые кислоты, полисахариды и смешанные биополимеры) и низкомолекулярные биологически активные вещества - витамины, гормоны, антибиотики, токсины и так далее, а также синтетические аналоги природных соединений, лекарственные препараты, пестициды и др.
Биоорганическая химия сформировалась как самостоятельная область во 2-й половине 20 века на стыке биохимии и органической химии на основе традиционной химии природных соединений. Её становление связано с именами Л. Полинга (открытие α-спирали и β-структуры как главных элементов пространственной структуры полипептидной цепи в белках), А. Тодда (выяснение химического строения нуклеотидов и первый синтез динуклеотида), Ф. Сенгера (разработка метода определения аминокислотной последовательности в белках и расшифровка с его помощью первичной структуры инсулина), В. Дю Виньо (выделение, установление структуры и химический синтез пептидных гормонов - окситоцина и вазопрессина), Д. Бартона и В. Прелога (конформационный анализ), Р. Вудворда (полный химический синтез многих сложных природных соединений, в том числе резерпина, хлорофилла, витамина В 12) и др.; в СССР огромную роль сыграли работы Н. Д. Зелинского, А. Н. Белозерского, И. Н. Назарова, Н. А. Преображенского и др. Инициатором исследований по биоорганической химии в СССР в начале 1960-х годов явился М. М. Шемякин. Им, в частности, были начаты работы (впоследствии получившие широкое развитие) по изучению циклических депсипептидов, выполняющих функцию ионофоров. Лидером отечественной биоорганической химии в 1970-80-х годах стал Ю.А. Овчинников, под руководством которого было установлено строение десятков белков, в том числе мембранных (впервые) - бактериородопсина и зрительного родопсина быка.
К основным направлениям биоорганической химии относятся:
1. Разработка методов выделения и очистки природных соединений. При этом для контроля за степенью очистки часто используют специфическую биологическую функцию изучаемого вещества (например, чистоту антибиотика контролируют по его антимикробной активности, гормона - по его влиянию на определённый биологический процесс и так далее). При разделении сложных природных смесей часто применяют методы высокоэффективной жидкостной хроматографии и электрофореза. С конца 20 века вместо поиска и выделения отдельных компонентов проводят тотальный скрининг биологических образцов на максимально возможное число компонентов того или иного класса соединений (смотри Протеомика).
2. Определение структуры изучаемых веществ. Под структурой понимают не только установление природы и порядок связи атомов в молекуле, но и их пространственное расположение. Для этого используют различные методы, в первую очередь химические (гидролиз, окислительное расщепление, обработка специфическими реагентами), позволяющие получать более простые вещества с известной структурой, по которым реконструируют структуру исходного вещества. Широко применяют автоматические устройства, обеспечивающие быстрое решение стандартных задач, особенно в химии белков и нуклеиновых кислот: анализаторы для количественного определения аминокислотного и нуклеотидного состава и секвенаторы для выяснения последовательности аминокислотных остатков в белках и нуклеотидов в нуклеиновых кислотах. Важную роль при изучении структуры биополимеров играют ферменты, особенно те, которые специфически расщепляют их по строго определённым связям (например, протеиназы, катализирующие реакции расщепления пептидных связей по остаткам глутаминовой кислоты, пролина, аргинина и лизина, или рестриктазы, специфически расщепляющие фосфодиэфирные связи в полинуклеотидах). Сведения о строении природных соединений получают также с помощью физических методов исследования - главным образом масс-спектрометрии, ядерного магнитного резонанса и оптической спектроскопии. Повышение эффективности химических и физических методов достигается благодаря одновременному анализу не только природных соединений, но и их производных, содержащих характерные, специально вводимые группировки и меченые атомы (например, путём выращивания бактерий - продуцентов того или иного соединения на среде, содержащей предшественников этого соединения, обогащённых стабильными или радиоактивными изотопами). Достоверность данных, получаемых при изучении сложных белков, значительно повышается при одновременном исследовании строения соответствующих генов. Пространственную структуру молекул и их аналогов в кристаллическом состоянии исследуют методом рентгеноструктурного анализа. Разрешение в ряде случаев достигает значений менее 0,1 нм. Для растворов наиболее информативен метод ЯМР в сочетании с теоретическим конформационным анализом. Добавочную информацию дают оптические спектральные методы анализа (электронные и флуоресцентные спектры, спектры кругового дихроизма и др.).
3. Синтез как самих природных соединений, так и их аналогов. Во многих случаях химический или химико-ферментативный синтез является единственным способом получения нужного вещества в больших (препаративных) количествах. Для относительно простых низкомолекулярных соединений встречный синтез служит важным критерием правильности ранее определённой структуры. Созданы автоматические синтезаторы белков и полинуклеотидов, позволяющие значительно сокращать время синтеза; с их помощью синтезирован ряд белков и полинуклеотидов, содержащих несколько сотен мономерных звеньев. Химический синтез - основной способ получения лекарственных препаратов неприродного происхождения. В случае природных веществ он часто дополняет биосинтез или конкурирует с ним.
4. Установление клеточной и молекулярной мишени, на которую направлено действие биологически активного вещества, выяснение химического механизма его взаимодействия с живой клеткой и её компонентами. Понимание молекулярного механизма действия необходимо для продуктивного использования биомолекул, с их зачастую чрезвычайно высокой активностью (например, токсинов), в качестве инструментов исследования биологических систем; оно служит основой для направленного синтеза новых, практически важных веществ с заранее заданными свойствами. В ряде случаев (например, при изучении пептидов, влияющих на деятельность нервной системы) получаемые таким образом вещества обладают многократно усиленной, по сравнению с исходным природным прототипом, изменённой в нужном направлении активностью.
Биоорганическая химия тесно связана с решением практических задач медицины и сельского хозяйства (получение витаминов, гормонов, антибиотиков и других лекарственных средств, стимуляторов роста растений, регуляторов поведения животных, в том числе насекомых), химической, пищевой и микробиологической промышленности. В результате сочетания методов биоорганической химии и генетической инженерии стало возможным практическое решение проблемы промышленного получения сложных, биологически важных веществ белково-пептидной природы, включая такие высокомолекулярные, как инсулин человека, α-, β- и γ-интерфероны, гормон роста человека.
Лит.: Дюга Г., Пенни К. Биоорганическая химия. М., 1983; Овчинников Ю. А. Биоорганическая химия. М., 1996.
Биоорганическая химия – наука, изучающая строение и свойства веществ, участвующих в процессах жизнедеятельности, в непосредственной связи с познанием их биологических функций.
Биоорганическая химия наука изучающая строение и реакционную способность биологически значимых соединений. Предметом биоорганической химии являются биополимеры и биорегуляторы и их структурные элементы.
К биополимерам относятся белки, полисахариды (углеводы) и нуклеиновые кислоты. В эту группу также включают липиды, которые не являются ВМС, но в организме обычно связаны с другими биополимерами.
Биорегуляторы – это соединения, которые химически регулируют обмен веществ. К ним относятся витамины, гормоны, многие синтетические соединения, в том числе лекарственные вещества.
Биоорганическая химия базируется на идеях и методах органической химии.
Без знания общих закономерностей органической химии, сложно изучение биоорганической химии. Биоорганическая химия тесно связана с биологией, биологической химией, медицинской физикой.
Совокупность реакций, протекающих в условиях организма, называется метаболизмом.
Вещества, образующиеся в процессе метаболизма, называются – метаболитами.
Метаболизм имеет два направления:
Катаболизм – реакции распада сложных молекул на более простые.
Анаболизм - это процесс синтеза сложных молекул из более простых веществ с затратой энергии.
Термин биосинтез применяется по отношению к химической реакции IN VIVO (в организме), IN VITRO (вне организма)
Существуют антиметаболиты - конкуренты метаболитов в биохимических реакциях.
Сопряжение, как фактор повышения стабильности молекул. Взаимное влияние атомов в молекулах органических соединений и способы ее передачи
План лекции:
Сопряжение и его виды:
p, p - сопряжение,
r,p - сопряжение.
Энергия сопряжения.
Сопряженные системы с открытой цепью.
Витамин А, каротины.
Сопряжение в радикалах и ионах.
Сопряженные системы с замкнутой цепью. Ароматичность, критерии ароматичности, гетероциклические ароматические соединения.
Ковалентная связь: неполярная и полярная.
Индуктивный и мезомерный эффекты. ЭА и ЭД – заместители.
Основным типом химических связей в органической химии являются ковалентные связи. В органических молекулах атомы соединены s и p - связями.
Атомы в молекулах органических соединениях соединены ковалентными связями, которые называются s и p - связями.

Одинарная s - связь в SP 3 – гибридизованном состоянии характеризуется l – длиной (С-С 0,154 нм) Е-энергией (83 ккал/моль), полярностью и поляризуемостью. Например:

Двойная связь характерна ненасыщенным соединениям, в которых, кроме центровой s - связи, есть еще перекрывание перпендикулярное s - связи которая называется π-связью).
Двойные связи бывают локализованными, то есть электронная плотность охватывает только 2 ядра связываемых атомов.

Чаще всего мы с вами будем иметь дело с сопряженными системами. Если же двойные связи чередуются с одинарными связями (а в общем случае у атома соединенного с двойной связью, есть р-орбиталь, то р-орбитали соседних атомов могут перекрываться друг с другом, образуя общую p - электронную систему). Такие системы называются сопряженными или делокализованными . Например: бутадиен-1,3

p, p - сопряженные системы
Все атомы в бутадиене находятся в SP 2 – гибридизированном состоянии и лежат в одной плоскости (Рz – не гибрид орбитали). Рz – орбитали параллельны друг другу. Это создает условия их взаимного перекрывания. Перекрывание Рz орбитали происходит между С-1 и С-2 и С-3 и С-4, а также между С-2 и С-3, то есть возникает делокализованная ковалентная связь. Это находит отражение в изменении длин связей в молекуле. Длина связи между С-1 и С-2 увеличена, а между С-2 и С-3 укорочена, по сравнению с одинарной связью.
l-C -С, 154 нм l С=С 0,134 нм
l С-N 1,147 нм l С =O 0,121 нм

r, p - сопряжение
Примером р, π сопряженной системы может служить пептидная связь.

r, p - сопряженные системы
Двойная связь С=0 удлиняется до 0,124 нм против обычной длины 0,121, а связь С – N становится короче и становится равной 0,132 нм по сравнению с 0,147 нм в обычном случае. То есть процесс делокализации электронов приводит к выравниванию длин связей и снижению внутренней энергии молекулы. Однако ρ,p – сопряжение возникает в ациклических соединениях, не только когда чередуется = связи с одинарными С- С связями,а еще при чередовании с гетероатомом:

Рядом с двойной связью может находиться атом Х, имеющий свободную р- орбиталь. Чаще всего это гетероатомы О,N, S и их р-орбитали, взаимодействуют с p - связями, образуя р, p - сопряжение.
Например:
СН 2 = СН – О – СН = СН 2
Сопряжение может осуществляться не только в нейтральных молекулах, но и в радикалах и ионах:

Исходя из выше изложенного, в открытых системах сопряжение возникает при следующих условиях:
Все атомы, участвующие в сопряженной системе, находятся в SP 2 – гибридизованном состоянии.
Рz – орбитали всех атомов перпендикулярны плоскости s - скелета, то есть параллельны друг другу.
При образовании сопряженной многоцентровой системы происходит выравнивание длин связей. Здесь нет «чистых» одинарных и двойных связей.
Делокализация p-электронов в сопряженной системе сопровождается выделением энергии. Система переходит на более низкий энергетический уровень, становится более устойчивой, более стабильной. Так, образование сопряженной системы в случае бутадиена – 1,3 приводит к выбросу энергии в количестве 15 кДж/моль. Именно за счет сопряжения повышается устойчивость радикалов ионов аллильного типа и их распространенность в природе.
Чем длиннее цепь сопряжения, тем больше выброс энергии ее образования.
Это явление довольно широко распространено в биологически важных соединениях. Например:


С вопросами термодинамической устойчивости молекул, ионов, радикалов мы будем постоянно встречаться в курсе биоорганической химии, к которым относятся ряд ионов и молекул широко распространенных в природе. Например: 
Сопряженные системы с замкнутой цепью
Ароматичность. В циклических молекулах при определенных условиях может возникать сопряженная система. Примером p, p - сопряженной системы является бензол, где p - электронное облако охватывает атомы углерода, такая система называется – ароматической.
Выигрыш энергии за счет сопряжения в бензоле составляет 150,6 кДж/моль. Поэтому бензол устойчив термически до температуры 900 о С.
Наличие замкнутого электронного кольца доказано с помощью ЯМР. Если молекулу бензола поместить во внешнее магнитное поле, возникает индуктивный кольцевой ток.
Таким образом, критерием ароматичности, сформулированным Хюккелем является:
молекула имеет циклическое строение;
все атомы находятся в SP 2 – гибридизованном состоянии;
существует делокализиванная p - электронная система, содержащая 4n + 2 электронов, где n – число циклов.
Например: 
Особое место в биоорганической химии занимает вопрос ароматичности гетероциклических соединений .
В циклических молекулах, содержащих гетероатомы (азот, сера, кислород) единое p - электронное облако, образуется с участием р – орбиталей атомов углерода и гетероатома.
Пятичленные гетероциклические соединения

Ароматическая система образуется при взаимодействии 4-х р-орбиталей С и одной орбитали гетероатома, на котором находится 2 электрона. Шесть p - электронов образуют ароматический скелет. Такая сопряженная система является электронно избыточной. В пирроле атом N находится в SP 2 гибридизированном состоянии.
Пиррол входит в состав многих биологически важных веществ. Четыре пиррольных кольца образуют порфин – ароматическую систему с 26 p - электронами и высокой энергией сопряжения (840 кДж/моль)
Порфиновая структура входит в состав гемоглобина и хлорофилла 

Шестичленные гетероциклические соединения
Ароматическая система в молекулах этих соединений образуется при взаимодействии пяти р-орбиталей атомов углерода и одной р-орбитали атома азота. Два электрона на двух SP 2 – орбитали участвует в образовании s - связей с атомами углерода кольца. Р-орбиталь с одним электроном входит в ароматический скелет. SP 2 – орбиталь с неподеленной парой электронов лежит в плоскости s - скелета.
Электронная плотность в пиримидине смещена к N, то есть система обеднена p - электронами, она электронно дефицитна.
Многие гетероциклические соединения могут содержать один и более гетероатомов 
Ядра пиррола, пиримидина, пурина входят в состав многих биологически активных молекул.
Взаимное влияние атомов в молекулах органических соединений и способы его передачи
Как уже отмечалось, связи в молекулах органических соединений осуществляются за счет s и p связей, электронная плотность равномерно распределена между связанными атомами только тогда, когда эти атомы одинаковы или близки по электроотрицательности. Такие связи называются неполярными. 
CH 3 -CH 2 →CI полярная связь
Чаще в органической химии имеем дело с полярными связями.
Если электронная плотность смешена в сторону более электроотрицательного атома, то такая связь называется – полярной. Основываясь на значениях энергии связей, американский химик Л.Полинг предложил количественную характеристику электроотрицательности атомов. Ниже представлена шкала Полинга.
Na Li H S C J Br Cl N O F
0,9 1,0 2,1 2,52,5 2,5 2,8 3,0 3,0 3,5 4,0
Атомы углерода в разном состоянии гибридизации различаются по электроотрицательности. Поэтому s - связь между SP 3 и SP 2 гибридизованными атомами - полярна 
Индуктивный эффект
Передача электронной плотности по механизму электростатической индукции по цепи s - связей называется индукцией , эффект называется индуктивным и обозначается J. Действие J, как правило, затухает через три связи, однако близко расположенные атомы испытывают довольно сильное влияние находящегося рядом диполя.
Заместители, смещающие электронную плотность по цепи s - связей в свою сторону, проявляют -J – эффект, а наоборот +J эффект. 

Изолированная p - связь, а также единое p - электронное облако открытой или замкнутой сопряженной системы способна легко поляризоваться под влиянием ЭА и ЭД заместителей. В этих случаях индуктивный эффект передается на p - связь, поэтому обозначает Jp. 
Мезомерный эффект (эффект сопряжения)
Перераспределение электронной плотности в сопряженной системе под влиянием заместителя, являющегося участником этой сопряженной системы, называется мезомерным эффектом
(М-эффект). 
Для того, чтобы заместитель сам входил в сопряженную систему, он должен иметь либо двойную связь (p,p -сопряжение), либо гетероатомом с неподеленной парой электронов (r,p - сопряжение). М – эффект передается по сопряженной системе без затухания.
Заместители, понижающие электронную плотность в сопряженной системе (смещенная электронная плотность в свою сторону) проявляют -М-эффект,а заместители, повышающие электронную плотность в сопряженной системе проявляют+М-эффект.
Электронные эффекты заместителей 
Реакционная способность органических веществ в значительной степени зависит от характера действия J и M эффектов. Знание теоретических возможностей действия электронных эффектов позволяет предсказать ход тех или иных химических процессов.
Кислотно-основные свойства органических соединений Классификация органических реакций.
План лекции
Понятие субстрата, нуклеофила, электрофила.
Классификация органических реакций.
обратимые и необратимые
радикальные, электрофильные, нуклеофильные, синхронные.
моно- и бимолекулярные
реакции замещения
реакции присоединения
реакции элиминирования
окисление и восстановление
кислотно-основные взаимодействия
Реакции региоселективные, хемоселективные, стереоселективные.
Реакции электрофильного присоединения. Правило Морковникова, антиморковниковское присоединение.
Реакции электрофильного замещения: ориентанты 1-го и 2-го рода.
Кислотно-основные свойства органических соединений.
кислотность и основность по Бренстеду
кислотность и основность по Льюису
Теория жестких и мягких кислит и оснований.
Классификация органических реакций
Систематизация органических реакций позволяет свести многообразие этих реакций к сравнительно не большому числу типов. Органические реакции можно классифицировать:
по направлению: обратимые и необратимые
по характеру изменения связей в субстрате и реагенте.
Субстрат – молекула, которая предоставляет атом углерода для образования новой связи
Реагент - действующее на субстрат соединение.
Реакции по характеру изменения связей в субстрате и реагенте можно разделить на:
радикальные R
электрофильные Е
нуклеофильные N (Y)
синхронные или согласованные 

Механизм реакций SR
Инициирование
Рост цепи
Обрыв цепи 
КЛАССИФИКАЦИЯ ПО КОНЕЧНОМУ РЕЗУЛЬТАТУ
Соответствие с конечным результатом реакции бывают:
А) реакции замещения
Б) реакции присоединения
В) реакции элиминирования
Г) перегруппировки
Д) окисление и восстановление
Е) кислотно-основные взаимодействия
Реакции также бывают:
Региоселективные – предпочтительно протекающие по одному из нескольких реакционных центров.
Хемоселективные – предпочтительное протекание реакции по одной из родственных функциональных групп.
Стереоселективные – предпочтительное образование одного из нескольких стереоизомеров.
Реакционная способность алкенов, алканов, алкадиенов, аренов и гетероциклических соединений
Основу органических соединений составляют углеводороды. Мы будем рассматривать лишь те реакции, осуществляемых в биологических условиях и соответственно не с самими углеводородами, а с участием углеводородных радикалов.
К ненасыщенным углеводородам мы относим алкены, алкадиены, алкины, циклоалкены и ароматические углеводороды. Объединяющее начало для них π – электронное облако. В динамических условиях также органические соединения склонны подвергаться атаке Е+
Однако, реакции взаимодействия для алкинов и аренов с реагенами приводит к разным результатам,так как, в этих соединениях разная природа π – электронного облака: локализованная и делокализиванная.
Рассмотрение механизмов реакций начнем с реакций А Е. Как нам известно, алкены взаимодействуют с 
Механизм реакции гидратации
По правилу Марковникова – присоединение к непредельным углеводородам несимметричного строения соединений с общей формулой НХ - атом водорода присоединяется к наиболее гидрогенизированому атому углерода,если заместитель ЭД. При антимарковниковском присоединении атом водорода присоединяется к наименее гидрогенизированному, если заместитель ЭА.
Реакции электрофильного замещения в ароматических системах имеют свои особенности. Первая особенность состоит в том, что для взаимодействия с термодинамически устойчивой ароматической системой требуются сильные электрофилы, которые, как правило, генерируются с помощью катализаторов.
Механизм реакции S E


ОРИЕНТИРУЮЩЕЕ ВЛИЯНИЕ
ЗАМЕСТИТЕЛЕЙ
Если в ароматическом ядре находится какой-либо заместитель, то он обязательно оказывает влияние на распределение электронной плотности кольца. ЭД – заместители (ориентанты 1-го ряда) СН 3 , ОН, ОR, NН 2 , NR 2 – облегчают замещение по сравнению с не замещенным бензолом и направляют входящую группу в орто- и пара- положение. Если ЭД заместители сильные, то не требуется катализатор эти реакции протекают в 3 стадии.
ЭА – заместители (ориентанты II-го рода) затрудняют реакции электрофильного замещения по сравнению с не замещенным бензолом. Реакции SЕ идет в более жестких условиях, входящая группа вступает в мета положение. К заместителям II рода относятся:
СООН, SО 3 Н, СНО, галогены и др.
Реакции SЕ характерны также для гетероциклических углеводородов. Пиррол, фуран, тиофен и их производные относятся к π- избыточным системам и достаточно легко вступает в реакции SЕ. Они легко галогенируются, алкилируются, ацилируются, сульфируются, нитрируются. При выборе реагентов необходимо учитывать их не стабильность в сильнокислотной среде т.е ацидофобность.
Пиридин и другие гетероциклические системы с пиридиновым атомом азота,являются π –не достаточными системами, они гораздо труднее вступают в реакции SЕ, при этом входящий электрофил занимает β-положение по отношению к атому азота. 
Кислотные и основные свойства органических соединений
Важнейшими аспектами реакционной способности органических соединений являются кислотно-основные свойства органических соединений.
Кислотность и основность также важные понятия, определяющие многие функциональные физико-химические и биологические свойства органических соединений. Кислотный и основной катализ является одной из наиболее распространенных ферментативных реакций. Слабые кислоты и основания – обычные компоненты биологических систем, играющие важную роль в метаболизме и его регуляции.
В органической химии существует несколько концепций кислот и оснований. Общепринятая в неорганической и органической химии теория кислот и оснований Бренстеда. Согласно Бренстеду, кислоты представляют вещества, способные отдать протон, а основания – вещества, способные присоединить протон. 
Кислотность по Бренстеду
В принципе, большинство органических соединений можно рассматривать как кислоты, поскольку в органических соединениях Н связан с С, N O S
Органические кислоты соответственно делятся на С – Н, N – Н, О – Н, S-Н – кислоты. 


Кислотность оценивается в виде Ка или - lg Ка = рКа, чем меньше рКа, тем сильнее кислота.
Количественная оценка кислотности органических соединений определена далеко не у всех органических веществ. Поэтому важно выработать умение проводить качественную оценку кислотных свойств различных кислотных центров. Для этого используют общий методический подход.
Сила кислоты определяется стабильностью аниона(сопряженного основания). Чем стабильнее анион, тем сильнее кислота.
Стабильность аниона определяется совокупностью ряда факторов:
электроотрицательностью и поляризуемостью элемента в кислотном центре.
степенью делокализации отрицательного заряда в анионе.
характером связанного с кислотным центром радикала.
сольватационными эффектами (влияние растворителя)
Рассмотрим последовательно роль всех этих факторов:
Влияние электроотрицательности элементов
Чем более электроотрицателен элемент, тем более делокализован заряд и тем стабильнее анион, тем сильнее кислота.
С (2,5) N (3,0) О(3.5) S (2,5)
Поэтому кислотность изменяется в ряду СН< NН < ОН Для SH – кислот преобладает другой фактор – поляризуемость. Атом серы больше по размеру и имеет вакантные d – орбитали. следовательно, отрицательный заряд способен делокализоваться в большом объеме, что приводит к большей стабильности аниона. Тиолы, как более сильные кислоты, реагируют с щелочами, а также с оксидами и солями тяжелых металлов, тогда, как спирты (слабые кислоты) способны реагировать только с активными металлами Относительно высокая кислотность толов используется в медицине, в химии лекарственных средств. Например: Применяют при отравлениях As, Hg, Cr, Bi, действие которых обусловлен связыванием металлов и выведением их из организма. Например: При оценке кислотности соединений с одинаковым атомом в кислотном центре определяющим фактором является делокализация отрицательного заряда в анионе. Стабильность аниона значительно повышается с появлением возможности делокализации отрицательного заряда по системе сопряженных связей. Значительное увеличение кислотности в фенолах, по сравнению со спиртами объясняется возможностью делокализации в ионах по сравнению с молекулой. Высокая кислотность карбоновых кислот обусловлена резонансной стабильностью карбоксилат аниона Делокализация заряда способствует наличие электроноакцепторных заместителей (ЭА) они стабилизируют анионы, тем самым увеличивают кислотность. Например, введение в молекулу ЭА заместителя Влияние заместителя и растворителя a - оксикислоты более сильные кислоты, чем соответствующие карбоновые кислоты. ЭД – заместители наоборот понижают кислотность. Растворители оказывают большее влияние на стабилизацию аниона, как правило лучше сольватируются небольшие ионы с низкой степенью делокализации заряда. Влияние сольватации можно проследить например в ряду: Если атом в кислотном центре несет положительный заряд это приводит к усилению кислотных свойств. Вопрос к аудитории: какая кислота – уксусная или пальмитиновая С 15 Н 31 СООН – должна иметь меньшее значение рКа? Если атом в кислотном центре несет положительных заряд, это приводит к усилению кислотных свойств. Можно отметить сильную СН – кислотность σ – комплекса, образующегося в реакции электрофильного замещения. Основность по Бренстеду Для того, чтобы образовать связь с протоном, необходимо не поделенная электронная пару у гетероатома, либо быть анионами. Существуют п-основания и π-основания, где центром основности являются электроны локализованной π-связи или π-электроны сопряженной системы (π-компоненты) Сила основания зависит от тех же факторов, что и кислотность, но влияние их противоположно. Чем больше электроотрицательность атома, тем прочнее он удерживает неподеленную пару электронов, и тем менее доступна она для связи с протоном. Тогда в целом сила n-оснований с одинаковым заместителем изменяется в ряду: Наибольшую основность из органических соединений проявляют амины и спирты: Соли органических соединений с минеральными кислотами хорошо растворимы. Многие лекарственные средства используют в виде солей. Кислотно-основной центр в одной молекуле(амфотерность)
Водородные связи как кислотно-основное взаимодействие Для всех α – аминокислот является преобладание катионных форм в сильнокислых и анионных в сильнощелочных средах. Наличие слабых кислотных и основных центров приводит к слабым взаимодействиям – водородным связям. Например: имидазол при небольшой молекулярной массе имеет высокую температуру кипения за счет наличия водородных связей. Дж. Льюисом предложена более общая теория кислот и оснований, определяющаяся на строении электронных оболочек. Кислотами по Льюису могут быть атом, молекула или катион, обладающие вакантной орбиталью, способное принимать пару электронов с образованием связи. Представителями кислот Льюиса служат галогениды элементов II и III групп периодической системы Д.И. Менделеева. Основания Льюиса атом, молекула или анион способный предоставлять пару электронов. К основаниям Льюиса относятся амины, спирты, простые эфиры, тиолы, тиоэфиры и содержащие π-связи соединения. Например, приведенное ниже взаимодействие можно представить как взаимодействие кислот и оснований Льюиса Важным следствие теории Льюиса является то, что любое органическое вещество можно представить как кислотно-основной комплекс. В органических соединениях внутримолекулярные водородные связи возникают значительно реже, чем межмолекулярные, но также имеют место в биоорганических соединениях и их можно рассматривать как кислотно-основные взаимодействия. Понятие «жесткие» и «мягкие» не тождественны сильным и слабым кислотам и основаниям. Это независимые две характеристики. Суть ЖКМО состоит в том, что жесткие кислоты реагируют с жесткими основаниями и мягкие кислоты реагируют с мягкими основаниями. В соответствии с принципом жестких и мягких кислот и оснований (ЖМКО) Пирсона кислоты Льюиса делятся на жесткие и мягкие. Жесткие кислоты- акцепторные атомы с малым размером,большим положительным зарядом, большой электроотрицательностью и низкой поляризуемостью. Мягкие кислоты- акцепторные атомы большого размера с малым положительным зарядом, с небольшой электроотрицательностью и высокой поляризуемостью
. Суть ЖКМО состоит в том, что жесткие кислоты реагируют с жесткими основаниями и мягкие кислоты реагируют с мягкими основаниями. Например: Окисление и восстановление органических соединений Окислительно-восстановительные реакции занимают важнейшее значение для процессов жизнедеятельности. С их помощью организм удовлетворяет свои энергетические потребности, поскольку при окислении органических веществ происходит высвобождение энергии. С другой стороны эти реакции служат для превращения пищи в компоненты клетки. Реакции окисления способствуют детоксикации и выведению лекарственных средств из организма. Окисление – процесс удаления водорода с образованием кратной связи или новых более полярных связей Восстановление – процесс обратный окислению. Окисление органических субстратов протекает тем легче, чем сильнее его тенденция к отдаче электронов. Окисление и восстановление необходимо рассматривать по отношению к определенным классам соединений. Окисление С – Н связей (алканов и алкилов) При полном сгорании алканов образуется СО 2 и Н 2 О при этом выделяется тепло. Другие пути их окисления и восстановления можно представить следующими схемами: Окисление насыщенных углеводородов протекает в жестких условиях (хромовая смесь горячая) более мягкие окислители не действуют на них. Промежуточными продуктами окисления являются спирты, альдегиды, кетоны, кислоты. Гидропероксиды R – О – ОН важнейшие промежуточные продукты окисления С – Н связей в мягких условиях, в частности in vivo Важной реакцией окисления С – Н связей в условиях организма является ферментативное гидроксилирование. Примером может быть получение спиртов при окислении пищи. За счет молекулярного кислорода и его активных форм. осуществляется в in vivo. Перекись водорода может служить в организме гидроксилирующим агентом. Избыток перекиси должен разлагаться с помощью каталазы на воду и кислород. Окисление и восстановление алкенов можно представить следующими превращениями: Восстановление алкенов Окисление и восстановление ароматических углеводородов Бензол чрезвычайно тяжело окисляется даже в жестких условиях по схеме: Способность к окислению заметно увеличивается от бензола к нафталину и далее к антрацену. ЭД- заместители облегчают окисление ароматических соединений. ЭА – затрудняют окисление. Восстановление бензола. С 6 Н 6 + 3Н 2 Ферментативное гидроксилирование ароматических соединений Окисление спиртов По сравнению с углеводородами, окисление спиртов осуществляется в более мягких условиях Важнейшей реакцией диолов в условиях организма является превращения в системе хинон-гидрохинон Перенос электронов от субстрата к кислороду осуществляется в метахондриях. Окисление и восстановление альдегидов и кетонов Один из наиболее легко окисляющийся классов органических соединений 2Н 2 С = О + Н 2 О СН 3 ОН + НСООН особенно легко протекает на свету Окисление азотсодержащих соединений Амины окисляются достаточно легко конечными продуктами окисления являются нитросоединения Исчерпывающее восстановление азотсодержащих веществ приводит к образованию аминов. Окисление аминов в in vivo Окисление и восстановление тиолов Cравнительная характеристика О-В свойств органических соединений. Наиболее легко окисляются тиолы и 2-х-атомные фенолы. Достаточно легко окисляются альдегиды. Труднее окисляются спирты, причем первичные легче, чем вторичные, третичные. Кетоны устойчивы к окислению или окисляются с расщеплением молекулы. Алкины окисляются легко даже при комнатной температуре. Наиболее трудно окисляются соединения, содержащие атомы углерода в Sр3-гибридизированом состоянии, то есть насыщенные фрагменты молекул. ЭД – заместители облегчают окисление ЭА – затрудняют окисление. Специфические свойства поли- и гетерофункциональных соединений. План лекции Поли- и гетерофункциональность, как фактор повышающий реакционную способность органических соединений. Специфические свойства поли- и гетерофункциональных соединений: амфотерность образование внутримолекулярных солей. внутримолекулярная циклизация γ, δ, ε – гетерофункциональных соединений. межмолекулярная циклизация (лактиды и декетопипирозины) хелатообразование. реакции элиминирования бета – гетерофункциональных соединений. таутомерия кето–енольная. Фосфоенолпируват, как макроэргическое соединение. декарбоксилирование. стереоизомерия Поли-и гетерофункциональность, как причина появления специфических свойств у гидрокси-, амино- и оксокислот. Наличие в молекуле нескольких одинаковых или разных функциональных групп составляет характерную черту биологически важных органических соединений. В молекуле может быть две и более гидроксильных групп, аминогрупп, карбоксильных групп. Например: Важную группу веществ участников жизнедеятельности составляют гетерофункциональные соединения, имеющие попарное сочетание разных функциональных групп. Например: В алифатических соединениях все приведённые функциональные группы проявляют ЭА характер. За счёт влияния друг на друга у них взаимно усиливается реакционная способность. Например, в оксокислотах электрофильность усиливается каждого из двух карбонильных атомов углерода под влиянием -J другой функциональной группы, что ведёт к более легкому восприятию атаки нуклеофильными реагентами. Поскольку I эффект затухает через 3–4 связи, то важным обстоятельством является близость расположения функциональных групп в углеводородной цепи. Гетерофункциональные группы могут находится у одного и того же атома углерода (α – расположение), или у разных атомов углерода как соседних(β расположение), так и более удалённых друг от друга (γ, дельта, эпсилон) расположения. Каждая гетерофункциональная группа сохраняет собственную реакционную способность, точнее гетерофункциональные соединения вступают как бы в «двойное» число химических реакций. При достаточном близком взаимном расположении гетерофункциональных групп происходит взаимное усиление реакционной способности каждой из них. При одновременном присутствии в молекуле кислотной и основной групп, соединение становятся амфотерным. Например: аминокислоты. Взаимодействие гетерофункциональных групп
В молекуле герофункциональных соединений могут содержатся группы, способные к взаимодействию друг с другом. Например, в амфотерных соединениях, как в α- аминокислотах, возможно образование внутренних солей. По этому все α – аминокислоты встречаются в виде биополярных ионов и хорошо растворимы в воде. Кроме кислотно–основных взаимодействий становятся возможны и другие виды химических реакций. Например, реакции S N у SP 2 гибрид атома углерода в карбонильной группе за счёт взаимодействия со спиртовой группой образование сложных эфиров, карбоксильной группы с аминогруппой (образование амидов). В зависимости от взаимного расположения функциональных групп эти реакции могут протекать как внутри одной молекулы (внутримолекулярные), так и между молекулами (межмолекулярные). Поскольку в результате реакции образуется циклические амиды, сложные эфиры. то определяющим фактором становится термодинамическая устойчивость циклов. В связи с этим конечный продукт, как правило, содержит шестичленный или пятичленный циклы. Чтобы при внутримолекулярном взаимодействии образовался в пяти или шестичленный сложноэфирный (амидный) цикл, гетерофункциональное соединение должно иметь в молекуле гамма или сигма расположение. Тогда в кл Химия
- наука о строении, свойствах веществ, их превращениях и сопровождающих явлениях.
Задачи:
1. Исследование строения вещества, развитие теории строения и свойств молекул и материалов. Важно установление связи между строением и разнообразными свойствами веществ и на этой основе построение теорий реакционной способности вещества, кинетики и механизма химических реакций и каталитических явлений. 2. Осуществление направленного синтеза новых веществ с заданными свойствами. Здесь также важно найти новые реакции и катализаторы для более эффективного осуществления синтеза уже известных и имеющих промышленное значение соединений. 3. Традиционная задача химии приобрела особое значение. Оно связано как с увеличением числа химических объектов и изучаемых свойств, так и с необходимостью определения и уменьшения последствий воздействия человека на природу. Химия является общетеоретической дисциплиной. Она призвана дать студентам современное научное представление о веществе как одном из видов движущейся материи, о путях, механизмах и способах превращения одних веществ в другие. Знание основных химических законов, владение техникой химических расчетов, понимание возможностей, предоставляемых химией с помощью других специалистов, работающих в отдельных и узких ее областях, значительно ускоряют получение нужного результата в различных сферах инженерной и научной деятельности. Химическая отрасль - одна из важнейших отраслей промышленности в нашей стране. Производимые ею химические соединения, различные композиции и материалы применяются повсюду: в машиностроении, металлургии, сельском хозяйстве, строительстве, электротехнической и электронной промышленности, связи, транспорте, космической технике, медицине, быту, и др. Главными направлениями развития современной химической промышленности являются: производство новых соединений и материалов и повышение эффективности существующих производств. В медицинском вузе студенты изучают общую, биоорганическую, биологическую химию, а также клиническую биохимию. Знания студентами комплекса химических наук в их преемственности и взаимосвязи дают большую возможность, больший простор в исследовании и практическом использовании различных явлений, свойств и закономерностей, способствует развитию личности. Специфическими особенностями изучения химических дисциплин в медицинском вузе являются: · взаимозависимость между целями химического и медицинского образования; · универсальность и фундаментальность данных курсов; · особенность построения их содержания в зависимости от характера и общих целей подготовки врача и его специализации; · единство изучения химических объектов на микро- и макроуровнях с раскрытием разных форм их химической организации как единой системы и проявляемых ею разных функций (химических, биологических, биохимических, физиологических и др.) в зависимости от их природы, среды и условий; · зависимость от связи химических знаний и умений с реальной действительностью и практикой, в том числе медицинской, в системе «общество - природа - производство - человек», обусловленных неограниченными возможностями химии в создании синтетических материалов и их значением в медицине, развитием нанохимии, а также в решении экологических и многих других глобальных проблем человечества. 1. Взаимосвязь между процессами обмена веществ и энергии в организме
Процессы жизнедеятельности на Земле обусловлены в значительной мере накоплением солнечной энергии в биогенных веществах - белках, жирах, углеводах и последующими превращениями этих веществ в живых организмах с выделением энергии. Особенно отчетливо понимание взаимосвязи химических превращений и энергетических процессов в организме было осознано после работы А. Лавуазье (1743-1794) и П. Лапласа (1749- 1827).
Они прямыми калориметрическими измерениями показали, что энергия, выделяемая в процессе жизнедеятельности, определяется окислением продуктов питания кислородом воздуха, вдыхаемым животными. Обмен веществ и энергии - совокупность процессов превращения веществ и энергии, происходящих в живых организмах, и обмен веществами и энергией между организмом и окружающей средой. Обмен веществ и энергии является основой жизнедеятельности организмов и принадлежит к числу важнейших специфических признаков живой материи, отличающих живое от неживого. В обмене веществ, или метаболизме, обеспеченном сложнейшей регуляцией на разных уровнях, участвует множество ферментных систем. В процессе обмена поступившие в организм вещества превращаются в собственные вещества тканей и в конечные продукты, выводящиеся из организма. При этих превращениях освобождается и поглощается энергия. С развитием в XIX-XX вв. термодинамики - науки о взаимопревращениях теплоты и энергий - стало возможно количественно рассчитывать превращение энергии в биохимических реакциях и предсказывать их направление. Обмен энергии может осуществляться передачей теплоты или совершением работы. Однако живые организмы не находятся в равновесии с окружающей средой и поэтому могут быть названы неравновесными открытыми системами. Тем не менее при наблюдении в течение определенного отрезка времени в химическом составе организма видимых изменений не происходит. Но это не значит, что химические вещества, составляющие организм, не подвергаются никаким превращениям. Напротив, они постоянно и достаточно интенсивно обновляются, о чем можно судить по скорости включения в сложные вещества организма стабильных изотопов и радионуклидов, вводимых в клетку в составе более простых веществ-предшественников. Между обменом веществ и обменом энергии существует одно принципиальное различие
. Земля не теряет и не получает сколько-нибудь заметного количества вещества. Вещество в биосфере обменивается по замкнутому циклу и т.о. используется многократно. Обмен энергией осуществляется иначе. Она не циркулирует по замкнутому циклу, а частично рассеивается во внешнее пространство. Поэтому для поддержания жизни на Земле необходим постоянный приток энергии Солнца. За 1 год в процессе фотосинтеза на земном шаре поглощается около 10 21 кал
солнечной энергии. Хотя она составляет лишь 0,02% всей энергии Солнца, это неизмеримо больше, чем та энергия, которая используется всеми машинами, созданными руками человека. Столь же велико количество участвующего в кругообороте вещества. 2. Химическая термодинамика как теоретическая основа биоэнергетики. Предмет и методы химической термодинамики
Химическая термодинамика
изучает переходы химической энергии в другие формы - тепловую, электрическую и т. п., устанавливает количественные законы этих переходов, а также направление и пределы самопроизвольного протекания химических реакций при заданных условиях. Термодинамический метод основан на ряде строгих понятий: «система», «состояние системы», «внутренняя энергия системы», «функция состояния системы». Объектом
изучения в термодинамике является система Одна и та же система может находиться в различных состояниях. Каждое состояние системы характеризуется определенным набором значений термодинамических параметров. К термодинамическим параметрам относятся температура, давление, плотность, концентрация и т. п. Изменение хотя бы только одного термодинамического параметра приводит к изменению состояния системы в целом. Термодинамическое состояние системы называют равновесным, если оно характеризуется постоянством термодинамических параметров во всех точках системы и не изменяется самопроизвольно (без затраты работы). Химическая термодинамика изучает систему в двух равновесных состояниях (конечном и начальном) и на этом основании определяет возможность (или невозможность) самопроизвольного течения процесса при заданных условиях в указанном направлении. Термодинамика изучает
взаимные превращения различных видов энергии, связанные с переходом энергии между телами в форме теплоты и работы. Термодинамика базируется на двух основных законах, получивших название первого и второго начал термодинамики. Предметом изучения
в термодинамике является энергия и законы взаимных превращений форм энергии при химических ре акциях, процессах растворения, испарения, кристаллизации. Хими́ческая термодина́мика - раздел физической химии, изучающий процессы взаимодействия веществ методами термодинамики. 3. Термодинамические системы: изолированные, закрытые, открытые, гомогенные, гетерогенные. Понятие о фазе.
Система
– это совокупность взаимодействующих веществ, мысленно или фактически обособленная от окружающей среды (пробирка, автоклав). Химическая термодинамика рассматривает переходы из одного состояния в другое, при этом могут изменяться или оставаться постоянными некоторые параметры
: · изобарические
– при постоянном давлении; · изохорические
– при постоянном объеме; · изотермические
– при постоянной температуре; · изобарно - изотермические
– при постоянном давлении и температуре и т.д. Термодинамические свойства системы можно выразить с помощью нескольких функций состояния системы
, называемых характеристическими функциями
: внутреннейэнергииU
, энтальпии
H
, энтропии
S
, энергии Гиббса
G
, энергии Гельмгольца
F
.
Характеристические функции обладают одной особенностью: они не зависят от способа (пути) достижения данного состояния системы. Их значение определяется параметрами системы (давлением, температурой и др.) и зависит от количества или массы вещества, поэтому принято относить их к одному молю вещества.
По способу передачи энергии, вещества и информации
между рассматриваемой системы и окружающей средой термодинамические системы классифицируются: 1. Замкнутая (изолированная) система
- это система в которой нет обмена с внешними телами ни энергией, ни веществом (в том числе и излучением) , ни информацией. 2. Закрытая система
- система в которой есть обмен только с энергией. 3. Адиабатно изолированная система -
это система в которой есть обмен энергией только в форме теплоты. 4. Открытая система
- это система, которая обменивается и энергией, и веществом, и информацией. Классификация систем
: 5.Первое начало термодинамики. Внутренняя энергия. Изобарный и изохорный тепловые эффекты
.
Первое начало термодинамики
- один из трёх основных законов термодинамики, представляет собой закон сохранения энергии для термодинамических систем. Первое начало термодинамики было сформулировано в середине XIX века в результате работ немецкого учёного Ю. Р. Майера, английского физика Дж. П. Джоуля и немецкого физика Г. Гельмгольца. Согласно первому началу термодинамики, термодинамическая система может совершать работу только за счёт своей внутренней энергии или каких-либо внешних источников энергии
. Первое начало термодинамики часто формулируют как невозможность существования вечного двигателя первого рода, который совершал бы работу, не черпая энергию из какого-либо источника. Процесс, протекающий при постоянной температуре, называется изотермическим
, при постоянном давлении - изобарическим
, при постоянном объеме – изохорическим.
Если во время процесса система изолирована от внешней среды таким образом, что исключен теплообмен со средой, процесс называют адиабатическим.
Внутренняя энергия системы.
При переходе системы из одного состояния в другое изменяются некоторые ее свойства, в частности внутренняя энергия U.
Внутренняя энергия системы представляет собой ее полную энергию, которая складывается из кинетической и потенциальной энергий молекул, атомов, атомных ядер и электронов. Внутренняя энергия включает в себя энергию поступательного, вращательного и колебательного движений, а также потенциальную энергию, обусловленную силами притяжения и отталкивания, действующими между молекулами, атомами и внутриатомными частицами. Она не включает потенциальную энергию положения системы в пространстве и кинетическую энергию движения системы как целого. Внутренняя энергия является термодинамической функцией состояния системы. Это значит, что всякий раз, когда система оказывается в данном состоянии, ее внутренняя энергия принимает определенное присущее этому состоянию значение.
∆U = U 2 - U 1 где U 1 и U 2 - внутренняя энергия системы в
конечном и начальном состояниях cсоответственно. Первый закон термодинамики.
Если система обменивается с внешней средой тепловой энергией Q и механической энергией (работой) А, и при этом переходит из состояния 1 в состоянии 2, количество энергии, которое выделится или поглощается системой форм теплоты Q или работой А равно полной энергии системы при переходе из одного состояния в другое и записывается. План 1. Предмет и значение биоорганической химии 2. Классификация и номенклатура органических соединений 3. Способы изображения органических молекул 4. Химическая связь в биоорганических молекулах 5. Электронные эффекты. Взаимное влияние атомов в молекуле 6. Классификация химических реакций и реагентов 7. Понятие о механизмах химических реакций 2
Предмет биоорганической химии 3 Биоорганическая химия самостоятельный раздел химической науки, что изучает строение, свойства и биологические функции химических соединений органического происхождения, которые принимают участие в обмене веществ живых организмов.
Обектами изучения биоорганической химии являются низкомолекулярные биомолекулы и биополимеры (белки, нуклеиновые кислоты и полисахариды), биоре-гуляторы (ферменты, гормоны, витамины и другие), природные и синтетические физиологически активные соединения, в том числе лекарственные средства и вещества с токсичным действием. Биомолекулы - биоорганические соединения, которые входят в состав живых организмов и специализированные для образования клеточных структур и участия в биохимических реакциях, составляют основу обмена веществ (метаболизм) и физиологических функций живых клеток и многоклеточных организмов в целом. 4 Классификация биоорганических соединений
Обмен веществ - совокупность химических реакций, которые протекают в организме (in vivo). Обмен веществ называют также метаболизмом. Метаболизм может происходить в двух направлениях – анаболизм и катаболизм. Анаболизм - это синтез в организме сложных веществ из сравнительно простых. Он протекает с затратой энергии (эндотермический процесс). Катаболизм - напротив, распад сложных органических соединений на более простые. Проходит он с выделением энергии (экзотермический процесс). Метаболитические процессы проходят при участии ферментов. Ф е р м е н т ы исполняют в организме роль био катализаторов. Без ферментов биохимические процессы или совсем не протекали бы, или протекали бы очень медленно и организм не смог бы поддерживать жизнь. 5
Биоэлементы. В состав биоорганических соединений, кроме атомов углерода (С), которые составляют основу любой органической молекулы, входят также водород (Н), кислород (О), азот (N), фосфор (Р) и сера (S). Эти биоэлементы (органогены) сконцентрированы в живых организмах в количестве, что свыше 200 раз превышает их содержание в объектах неживой природы. Отмеченные элементы составляют свыше 99% элементного состава биомолекул. 6
Биоорганическая химия возникла из недр органической химии и базируется на ее идеях и методах. В истории развития для органической химии отведены такие этапы: эмпирический, аналитический, структурный и современный. Период от первого знакомства человека с органическими веществами до конца XVІІІ века считается эмпирическим. Основной итог этого периода – люди осознали значение элементного анализа и установление атомных и молекулярных масс. Теория витализма - жизненной силы (Берцелиус). До 60-х годов XІX века продолжался аналитический период. Он ознаменовался тем, что с конца первой четверти XІX века были сделаны ряд перспективных открытий, которые нанесли сокрушительный удар по виталистической теории. Первым в этом ряду был ученик Берцелиуса, немецкий химик Велер. Он осуществил ряд открытий в 1824 г. – синтез щавелевой кислоты из дициана: (CN) 2 НООС – СООН р. – синтез мочевины из аммония цианата: NH 4 CNO NH 2 – C – NH 2 О 8
В 1853 Ш. Жерар разработал «теорию типов» и и пользовал её для классификации органических соединений. Согласно Жерару, более сложные органические соединения могут быть произведены от следующих основных четырёх типов веществ: НННН тип ВОДОРОДА НННН O тип ВОДЫ Н Cl тип ХЛОРИСТОГО ВОДОРОДА ННHННH N тип АММИАКА С 1857 по предложению Ф. А. Кекуле углеводороды стали относить к типу метана ННHНННHН С 9
Основные положения теории строения органических соединений (1861) 1) атомы в молекулах соединены друг с другом химическими связями в соответствии с их валентностью; 2) атомы в молекулах органических веществ соединяются между собой в определенной последовательности, что обусловливает химическое строение (структуру) молекулы; 3) свойства органических соединений зависят не только от числа и природы входящих в их состав атомов, но и от химического строения молекул; 4) в органических молекулах существует взаимодействие между атомами, как связанными друг с другом, так и несвязанными; 5) химическое строение вещества можно определить в результате изучения его химических превращений и, наоборот, по строению вещества можно характеризовать его свойства. 10
Основные положения теории строения органических соединений (1861) Структурная формула это изображение последовательности связи атомов в молекуле. Брутто-формула – СН 4 О или CH 3 OH Структурная формула Упрощенные формулы строения иногда называют рациональными Молекулярная формула - формула органического соединения, которая указывает на количество атомов каждого элемента в молекуле. Например: С 5 Н 12 – пентан, С 6 Н 6 – бензин и т.д. 11
Етапы развития биоорганической химии Как отдельная область знаний, которая совмещает концептуальные принципы и методологию органической химии с одной стороны и молекулярной биохимии и молекулярной фармакологии с другой стороны, биоорганическая химия сформировалась в годах ХХ столетия на основании разработок химии природных веществ и биополимеров. Фундаментальное значение современная биоорганическая химия приобрела благодаря работам В. Стейна, С. Мура, Ф. Сенгера (анализ аминокислотного состава и определение первичной структуры пептидов и белков), Л. Полинга и Х. Астбери (выяснение строения -спирали и -структуры и их значение в реализации биологических функций белковых молекул), Е. Чаргаффа (расшифровывание особенностей нуклеотидного состава нуклеиновых кислот), Дж. Уотсона, Фр. Крика, М. Уилкинса, Р. Франклина (установление закономерностей пространственной структуры молекулы ДНК), Г. Корани (химический синтез гена) и т.д. 14
Классификация органических соединений по строению углеродного скелета и природе функциональной группы Огромное число органических соединений побудило химиков провести их классификацию. В основу классификации органических соединений положены два классификационных признака: 1. Строение карбонового скелета 2. Природа функциональных групп Классификация по способу строения карбонового скелета: 1. Ациклические (алканы, алкены, алкины, алкадиены); 2. Циклические 2.1. Карбоциклические (алициклические и ароматические) 2.2. Гетероциклические 15 Ациклические соединения называют еще алифатическими. К ним принадлежат вещества с незамкнутой углеродной цепью. Ациклические соединения делят на насыщенные (или предельные) С n H 2n+2 (алканы, парафины) и ненасыщенные (непредельные). К последним относят алкены С n H 2n, алкины С n H 2n -2, алкадиены С n H 2n -2.
16 Циклические соединения в составе своих молекул содержат кольца (циклы). Если в состав циклов входят лишь атомы углерода, то такие соединения называют карбоциклическими. В свою очередь карбоциклические соединения разделяют на алициклические и ароматические. К алициклическим углеводородам (циклоалканы) относят циклопропан и его гомологи - циклобутан, циклопентан, циклогексан и так далее. Если же в циклическую систему кроме углеводорода входят и другие элементы, то такие соединения относят к гетероциклическим.
Классификация по природе функциональной группы Функциональная группа – это атом или группа определенным образом связанных атомов, наличие которых в молекуле органического вещества определяет характерные свойства и его принадлежность к тому или другому классу соединений. По количеству и однородности функциональных групп органические соединения делят на моно-, поли- и гетерофункциональные. Вещества с одной функциональной группой называют монофунк-циональными, с несколькими одинаковыми функциональными группами полифункциональными. Соединения, содержащие несколько различных функциональных групп гетеро функциональными. Важно, что соединения одного класса объединены в гомологические ряды. Гомологический ряд это ряд органических соединений с одинаковыми функциональными группами и однотипным строением, каждый представитель гомологического ряда отличается от предыдущего на постоянную единицу (СН 2), которую называют гомологической разностью. Члены гомологического ряда называют гомологами. 17
Номенклатурные системы в органической химии – тривиальная, рациональная и международная (IUPAC) Химическая номенклатура совокупность названий индивидуальных химических веществ, их групп и классов, а также правила составления их названий.Химическая номенклатура совокупность названий индивидуальных химических веществ, их групп и классов, а также правила составления их названий. Тривиальная (историческая) номенклатура связана с процессом получения веществ (пирогаллол – продукт пиролиза галловой кислоты), источника происхождения, из которого получали (муравьиная кислота) и т.д. Тривиальные названия соединений широко применяют в химии природных и гетероциклических соединений (цитраль, гераниол, тиофен, пиррол, хинолин, и др.).Тривиальная (историческая) номенклатура связана с процессом получения веществ (пирогаллол – продукт пиролиза галловой кислоты), источника происхождения, из которого получали (муравьиная кислота) и т.д. Тривиальные названия соединений широко применяют в химии природных и гетероциклических соединений (цитраль, гераниол, тиофен, пиррол, хинолин, и др.). В основе рациональной номенклатуры используется принцип деления органических соединений на гомологические ряды. Все вещества в определенном гомологическом ряду рассматриваются как производные самого простого представителя данного ряда – первого или иногда второго. В частности, у алканов – метана, у алкенов – этилена и т.д.В основе рациональной номенклатуры используется принцип деления органических соединений на гомологические ряды. Все вещества в определенном гомологическом ряду рассматриваются как производные самого простого представителя данного ряда – первого или иногда второго. В частности, у алканов – метана, у алкенов – этилена и т.д. 18
Международная номенклатура (IUPAC). Правила современной номенклатуры были разработаны в 1957 году на ХІХ конгрессе Международного союза теоретической и прикладной химии (International Union of Pure and Applied Chemistry – IUPAC). Радикально-функциональная номенклатура. В основе этих названий лежит название функционального класса (спирт, эфир, кетон и др.), которому предшествуют названия углеводородных радикалов, например: алилхлорид, диэтиловый эфир, диметилкетон, пропиловый спирт и т.д. Заместительная номенклатура. Правила номенклатуры. Родоначальная структура - структурный фрагмент молекулы (молекулярный остов), лежащий в основе названия соединения, главная углеродная цепь атомов для алициклических соединений, для карбоциклических – цикл. 19
Химическая связь в органических молекулах Химическая связь – явление взаимодействия внешних электронных оболочек (валентных электронов атомов) и ядер атомов, обуславливающее существование молекулы или кристалла как целого. Как правило атом, принимая, отдавая электрон или образуя общую электронную пару, стремится приобрести конфигурацию внешней электронной оболочки аналогичную инертным газам. Для органических соединений характерны следующие типы химических связей: - ионная связь -ковалентная связь -донорно - акцепторная связь -водородная связь Также существуют некоторые другие типы химической связи (металлическая, одноэлектронная, двухэлектронная трехцентровая), однако в органических соединениях они практически не встречаются. 20
Типы связей в органических соединениях Наиболее характерной для органических соединений является ковалентная связь. Ковалентная связь - это взаимодействие атомов, которое реализуется посредством образования общей электронной пары. Данный тип связи образуется между атомами, которые имеют сравнимые значения электроотрицательности. Электроотрицательность - свойство атома, показывающее способность оттягивать к себе электроны от других атомов. Ковалентная связь может быть полярной или неполярной. Неполярная ковалентная связь возникает между атомами с одинаковым значением электроотрицательности
Типы связей в органических соединениях Ковалентная полярная связь образуется между атомами, которые имеют различные значения электроотрицательности. В данном случае связанные атомы приобретают частичные заряды δ+δ+ δ-δ- Особым подтипом ковалентной связи является донорно- акцепторная связь. Как и в предыдущих примерах данный тип взаимодействия обусловлен наличием общей электронной пары, однако последняя предоставляется одним из атомов образующих связь (донором) и принимается другим атомом (акцептором) 24
Типы связей в органических соединениях Ионная связь образуется между атомами, которые сильно отличаются по значениям электроотрицательности. В данном случае электрон менее электроотрицательного элемента (зачастую это металл) полностью переходит к более электроотрицательному элементу. Данный переход электрона обуславливает появление положительного заряда у менее электроотрицательного атома и отрицательного у более электроотрицательного. Таким образом, образуется два иона с противоположным зарядом, между которыми существует электровалентное взаимодействие. 25
Типы связей в органических соединениях Водородная связь является электростатическим взаимодействием между атомом водорода, который связан сильнополярной связью, и электронными парами кислорода, фтора, азота, серы и хлора. Данный тип взаимодействия является довольно слабым взаимодействием. Водородная связь может быть межмолекулярной и внутримолекулярной. Межмолекулярная водородная связь (взаимодействие между двумя молекулами этилового спирта) Внутримолекулярная водородная связь в салициловом альдегиде 26
Химическая связь в органических молекулах Современная теория химической связи основывается на квантово-механической модели молекулы как системы, состоящей из электронов и атомных ядер. Краеугольным понятием квантово-механической теории является атомная орбиталь. Атомная орбиталь – часть пространства, в котором вероятность нахождения электронов является максимальной. Связь, таким образом, может быть рассмотрена как взаимодействие («перекрывание») орбиталей, которые несут по одному электрону с противоположными спинами. 27
Гибридизация атомных орбиталей Согласно квантово-механической теории, количество образованных атомом ковалентных связей определяется количеством одноэлектронных атомных орбиталей (количеством неспаренных электронов). У атома углерода в основном состоянии всего два неспаренных электрона, однако возможный переход электрона с 2s на 2 рz обуславливает возможность образования четырех ковалентных связей. Состояние атома углерода, при котором он имеет четыре неспаренных электрона называется «возбужденным». Несмотря на то, что орбитали углерода являются неравноценными, известно, что возможно образование четырех эквивалентных связей вследствие гибридизации атомных орбиталей. Гибридизация – явление, при котором из нескольких разных по форме и близких по энергии орбиталей образуется такое же количество одинаковых по форме и количеству орбиталей. 28
Гибридные состояния атома углерода в органических молекулах ПЕРВОЕ ГИБРИДНОЕ СОСТОЯНИЕ Атом С находится в состоянии sp 3 –гибридизации, образует четыре σ-связи, формирует четыре гибридные орбитали, которые располагаются в форме тетраэдра (валентный угол) σ–связь 31
Гибридные состояния атома углерода в органических молекулах ВТОРОЕ ГИБРИДНОЕ СОСТОЯНИЕ Атом С находится в состоянии sp 2 –гибридизации, образует три σ- связи, формирует три гибридные орбитали, которые располагаются в форме плоского треугольника (валентный угол 120) σ–связи π–связь 32
Гибридные состояния атома углерода в органических молекулах ТРЕТЬЕ ГИБРИДНОЕ СОСТОЯНИЕ Атом С находится в состоянии sp–гибридизации, образует две σ- связи, формирует две гибридные орбитали, которые располагаются в линию (валентный угол 180) σ–связи π–связи 33
Характеристики химических связей Шкала ПОЛИНГА: F-4,0; O – 3,5; Cl – 3,0; N – 3,0; Br – 2,8; S – 2,5; C-2,5; H-2,1. разница 1,7 Характеристики химических связей Поляризуемость связи - смещение электронной плотности под действием внешних факторов. Поляризуемость связи - степень подвижности электронов. С увеличением атомного радиуса возростает поляризуемость электронов. Поэтому поляризуемость связи Карбон - галоген увеличивается следующим образом: C-F
Электронные эффекты. Взаимное влияние атомов в молекуле 39 По современным теоретическим представлениям, реакционная способность органических молекул предопределена смещением и подвижностью электронных облаков, которые образуют ковалентную связь. В органической химии различают два типа смещений электронов: а) электронные смещения, происходящие в системе -связей, б) электронные смещения, передающиеся системой -связей. В первом случае имеет место так называемый индуктивный эффект, во втором - мезомерный. Индуктивный эффект - перераспределение электронной плотности (поляризация), возникающее в результате разницы электроотрицательности между атомами молекулы в системе -связей. Через незначительную поляризуемость -связей индуктивний эффект быстро угасает и через 3-4 связи он почти не проявляется.
Электронные эффекты. Взаимное влияние атомов в молекуле 40 Понятие об индуктивном эффекте было введено К. Ингольдом, им же были введены обозначения: –I-эффект в случае понижения заместителем электронной плотности +I-эффект в случае повышения заместителем электронной плотности Положительный индуктивний эффект проявляют алкильные радикалы (СН 3, С 2 Н 5 - и т.д.). Все другие заместители, связанные с атомом углерода, проявляют отрицательный индуктивный эффект.
Электронные эффекты. Взаимное влияние атомов в молекуле 41 Мезомерным эффектом называют перераспределение электронной плотности вдоль сопряженной системы. К сопряженным системам принадлежат молекулы органических соединений, в которых чередуются двойные и одинарные связи или когда рядом с двойной связью размещен атом, имеющий на р-орбитали неподеленную пару электронов. В первом случае имеет место, - сопряжение, а во втором – р, -сопряжение. Сопряженные системы бывают с открытой и замкнутой цепью сопряжения. Примером таких соединений является 1,3-бутадиен и бензин. В молекулах этих соединений атомы углерода находятся в состоянии sp 2 -гибридизации и за счет негибридных p-орбиталей образуют -связи, которые взаимно перекрываются между собой и формируют единственное электронное облако, то есть имеет место сопряжение.
Электронные эффекты. Взаимное влияние атомов в молекуле 42 Существует два вида мезомерного эффекта – положительный мезомерный эффект (+М) и отрицательный мезомерный эффект (-М). Положительный мезомерный эффект проявляют заместители, предоставляющие p-электроны в сопряженную систему. К таковым относят: -O, -S -NН 2, -ОН, -OR, Hal (галогены) и другие заместители, которые имеют отрицательный заряд или неподеленную пару электронов. Отрицательный мезомерный эффект характерен для заместителей оттягивающих на себя -электронную плотность из сопряженной системы. К таковым относят заместители, имеющие кратные связи между атомами с разной электроотрицательностью: - N0 2 ; -SO 3 Н; >С=О; -СООН и другие. Мезомерный эффект графически отражается согнутой стрелкой, которая показывает направление смещения электронов В отличие от индукционного эффекта, мезомерный эффект не погасает. Он передается полностью по системе, независимо от длины цепи сопряжения.
С=О; -СООН и другие. Мезомерный эффект графически отражается согнутой стрелкой, которая показывает направление смещения электронов В отличие от индукционного эффекта, мезомерный эффект не погасает. Он передается полностью по системе, независимо от длины цепи сопряжения.">
Типы химических реакций 43 Химическую реакцию можно рассматривать как взаимо- действие реагента и субстрата. В зависимости от способа разрыва и образования химической связи в молекулах, органические реакции делят на: а) гомолитические б) гетеролитические в) молекулярные Гомолитические или свободно-радикальные реакции обусловлены гомолитическим разрывом связи, когда у каждого атома остается по одному электрону, то есть образуются радикалы. Гомолитический разрыв проис- ходит при высоких температурах, действии кванта света или катализе.
Гетеролитические или ионные реакции протекают таким образом, что пара связующих электронов остается около одного из атомов и образуются ионы. Частица с электронной парой называется нуклеофи- льной и имеет отрицательный заряд (-). Частица без электронной пары называется электрофи- льной и имеет положительный заряд (+). 44 Типы химических реакций
Механизм химической реакции 45 Механизмом реакции называют совокупность элементарных (простых) стадий, из которых состоит данная реакция. Механизм реакции чаще всего включает такие стадии: активация реагента с образованием электрофила, нуклеофила или свободного радикала. Для активации реагента нужен, как правило, катализатор. Во второй стадии происходит взаимодействие активированного реагента с субстратом. При этом образуются промежуточные частицы (интермедиаты). К последним принадлежат -комплексы, -комплексы (карбокатионы), карбанионы, новые свободные радикалы. На конечной стадии проходит присоединение или отщепление к (от) образованному во второй стадии интермедиату какой-то частицы с формированием конечного продукта реакции. Если реагент при активации генерирует нуклеофил, то это - нуклеофильные реакции. Их помечают буквой N -(в индексе). В случае, когда реагент генерирует электрофил, реакции принадлежат к электрофильным (Е). Аналогично можно сказать и о свободнорадикальных реакциях (R).
Нуклеофилы – реагенты, имеющие отрицательный заряд или обогащенный электронной плотностью атом: 1) анионы: OH -, CN -, RO -, RS -, Hal - и другие анионы; 2) нейтральные молекулы с неподеленными парами электронов: NH 3, NH 2 R, H 2 O, ROH и другие; 3) молекулы с избыточной электронной плотностью (имеющие - связи). Электрофилы – реагенты, имеющие положительный заряд или обедненный электронной плотностью атом: 1) катионы: Н + (протон), НSO 3 + (ион гидрогенсульфония), NO 2 + (ион нитрония), NO (ион нитрозония) и другие катионы; 2) нейтральные молекулы, имеющие вакантную орбиталь: AlCl 3, FeBr 3, SnCl 4, BF 4 (кислоты Льюиса), SO 3 ; 3) молекулы с обедненной электронной плотностью на атоме. 46
49
50
51
52
ЛЕКЦИЯ 1
Биоорганическая химия (БОХ), ее значение в медицине
БОХ – это наука, изучающая биологическую функцию органических веществ в организме. БОХ возникла во 2-ой половине ХХ века. Объектами ее изучения служат биополимеры, биорегуляторы и отдельные метаболиты. Биополимеры – высокомолекулярные природные соединения, которые являются основой всех организмов. Это пептиды, белки, полисахариды, нуклеиновые кислоты (НК), липиды и др. Биорегуляторы – соединения, которые химически регулируют обмен веществ. Это витамины, гормоны, антибиотики, алкалоиды, лекарственные препараты и др. Знание строения и свойств биополимеров и биорегуляторов позволяет познать сущность биологических процессов. Так, установление строения белков и НК позволило развить представления о матричном биосинтезе белка и роли НК в сохранении и передаче генетической информации. БОХ играет большую роль в установлении механизма действия ферментов, лекарств, процессов зрения, дыхания, памяти, нервной проводимости, мышечного сокращения и др. Основная проблема БОХ – это выяснение взаимосвязи структуры и механизма действия соединений. БОХ основана на материале органической химии. ОРГАНИЧЕСКАЯ ХИМИЯ
Это наука, изучающая соединения углерода. В настоящее время насчитывается ~ 16 млн. органических веществ. Причины многообразия органических веществ. 1. Соединения атомов С друг с другом и др. элементами периодической системы Д. Менделеева. При этом образуются цепи и циклы: Прямая цепь Разветвленная цепь Тетраэдрическая Плоскостная конфигурация конфигурация атома С атома С 2. Гомология – это существование веществ с близкими свойствами, где каждый член гомологического ряда отличается от предыдущего на группу 3. Изомерия – это существование веществ, имеющих одинаковый качественный и количественный состав, но различное строение. А.М. Бутлеров (1861) создал теорию строения органических соединений, которая и по сей день служит научной основой органической химии. Основные положения теории строения органических соединений: 1) атомы в молекулах соединены друг с другом химическими связями в соответствии с их валентностью; 2) атомы в молекулах органических соединений соединяются между собой в определенной последовательности, что обусловливает химическое строение молекулы; 3) свойства органических соединений зависят не только от числа и природы входящих в их состав атомов, но и от химического строения молекул; 4) в молекулах существует взаимное влияние атомов как связанных, так и непосредственно друг с другом не связанных; 5) химическое строение вещества можно определить в результате изучения его химических превращений и, наоборот, по строению вещества можно охарактеризовать его свойства. Рассмотрим некоторые положения теории строения органических соединений. Структурная изомерия
Она делится: 1) Изомерия цепи 2) Изомерия положения кратной связи и функциональных групп 3) Изомерия функциональных групп (межклассовая изомерия) Формулы Ньюмена
Циклогексан Форма «кресла» более энергетически выгодна, чем «ванна». Конфигурационные изомеры
Это стереоизомеры, молекулы которых имеют различное расположение атомов в пространстве без учета конформаций. По типу симметрии все стереоизомеры делятся на энантиомеры и диастереомеры. Энантиомеры (оптические изомеры, зеркальные изомеры, антиподы) – это стереоизомеры, молекулы которых относятся между собой как предмет и несовместимое с ним зеркальное отображение. Это явление наз-ся энантиомерией. Все химические и физические св-ва энантиомеров одинаковы, кроме двух: вращение плоскости поляризованного света (в приборе поляриметре) и биологическая активность. Условия энантиомерии: 1) атом С находится в состоянии sp 3 -гибридизации; 2) отсутствие всякой симметрии; 3) наличие асимметрического (хирального) атома С, т.е. атома, имеющего четыре
разных заместителя. Многие окси- и аминокислоты обладают способностью вращать плоскость поляризации луча света влево или вправо. Это явление наз-ся оптической активностью, а сами молекулы оптически активными. Отклонение луча света вправо отмечают знаком «+», влево – «–» и указывают угол вращения в градусах. Абсолютную конфигурацию молекул определяют сложными физико-химическими методами. Относительную конфигурацию оптически активных соединений определяют путем сравнения со стандартом глицеринового альдегида. Оптически активные вещ-ва, имеющие конфигурацию правовращающего или левовращающего глицеринового альдегида (М. Розанов, 1906), наз-ся вещ-вами D- и L-ряда. Равная смесь право- и левовращающих изомеров одного соединения наз-ся рацематом и оптически неактивна. Исследования показали, что знак вращения света нельзя связывать с принадлежностью вещ-ва к D- и L-рядам, его определяют только экспериментально в приборах – поляриметрах. Например, L-молочная к-та имеет угол вращения +3,8 о, D- молочная к-та - -3,8 о. Энантиомеры изображают с помощью формул Фишера. L-ряд D-ряд Среди энантиомеров могут быть симметричные молекулы, не обладающие оптической активностью, и наз-ся мезоизомерами. Рацемат – виноградная к-та Оптические изомеры, не являющиеся зеркальными изомерами, отличающиеся конфигурацией нескольких, но не всех асимметрических атомов С, обладающие различными физическими и химическими св-вами, наз-ся s-ди
-а
-стереоизомерами. p-Диастереомеры (геометрические изомеры) – это стереомеры, имеющие в молекуле p-связь. Они встречаются у алкенов, непредельных высших карбоновых к-т, непредельных дикарбоновых к-т Биологическая активность органических вещ-в связана с их строением. Например: Цис-бутендиовая к-та, Транс-бутендиовая к-та, малеиновая к-та – фумаровая к-та – не ядовита, очень ядовита содержится в организме Все природные непредельные высшие карбоновые к-ты являются цис-изомерами. ЛЕКЦИЯ 2
Сопряженные системы
В простейшем случае сопряженные системы – это системы с чередующимися двойными и одинарными связями. Они могут быть открытыми и закрытыми. Открытая система имеется в диеновых углеводородах (УВ). СН 2 = СН – СН = СН 2 Бутадиен-1, 3 СН 2 = СН – Сl
Здесь происходит сопряжение p-электронов с р-электронами. Этот вид сопряжения наз-ся р, p-сопряжением. Закрытая система имеется в ароматических УВ. Ароматичность
Это понятие, включающее различные свойства ароматических соединений. Условия ароматичности: 1) плоский замкнутый цикл, 2) все атомы С находятся в sp 2 – гибридизации, 3) образуется единая сопряженная система всех атомов цикла, 4) выполняется правило Хюккеля: “В сопряжении участвуют 4n+2 p-электронов, где n = 1, 2, 3... ” Простейший представитель ароматических УВ – бензол. Он удовлетворяет всем четырем условиям ароматичности. Правило Хюккеля: 4n+2 = 6, n = 1. Взаимное влияние атомов в молекуле В 1861 г русский ученый А.М. Бутлеров высказал положение: «Атомы в молекулах взаимно влияют друг на друга». В настоящее время это влияние передается двумя путями: индуктивным и мезомерным эффектами. Индуктивный эффект
Это передача электронного влияния по цепи s-связи. Известно, что связь между атомами с различной электроотрицательностью (ЭО) поляризована, т.е. смещена к более ЭО атому. Это приводит к появлению на атомах эффективных (реальных) зарядов (d). Такое электронное смещение наз-ся индуктивным и обозначается буквой I и стрелкой ®. Индуктивный эффект может быть положительным или отрицательным. Если заместитель Х притягивает электроны химической связи сильнее, чем атом Н, то он проявляет – I. I(Н) = О. В нашем примере Х проявляет – I. Если заместитель Х притягивает электроны связи слабее, чем атом Н, то он проявляет +I. Все алкилы (R = СН 3 -, С 2 Н 5 - и т.д.), Ме n + проявляют +I. Мезомерный эффект
Мезомерный эффект (эффект сопряжения) – это влияние заместителя, передаваемое по сопряженной системе p-связей. Обозначается буквой М и изогнутой стрелкой. Мезомерный эффект может быть «+» или «–». Выше было сказано, что имеется два вида сопряжения p, p и р, p. Заместитель, притягивающий электроны из сопряженной системы, проявляет –М и наз-ся электроноакцептором (ЭА). Это заместители, имеющие двой- ную связь и др. Заместитель, отдающий электроны в сопряженную систему, проявляет +М и наз-ся электронодонором (ЭД). Это заместители с одинарными связями, имеющие неподеленную электронную пару (и др.). Таблица 1 Электронные эффекты заместителей
ЛЕКЦИЯ 3
Кислотность и основность
Для характеристики кислотности и основности органических соединений применяют теорию Бренстеда. Основные положения этой теории: 1) Кислота – это частица, отдающая протон (донор Н +); основание – это частица, принимающая протон (акцептор Н +). 2) Кислотность всегда характеризуется в присутствии оснований и наоборот. А – Н + : В Û А – + В – Н + осн-ие к-та СН 3 СООН + НОН Û СН 3 СОО – + Н 3 О + К-та Осн-ие Сопряженное Сопряженная осн-ие к-та НNО 3 + СН 3 СООH Û СН 3 СООН 2 + + NО 3 - К-та Осн-ие Сопряженная Сопряженное к-та осн-ие Кислоты Бренстеда
3) К-ты Бренстеда делятся на 4 вида в зависимости от кислотного центра: SН к-ты (тиолы), ОН к-ты (спирты, фенолы, карбоновые к-ты), NН к-ты (амины, амиды), СН к-ты (УВ). В этом ряду сверху вниз кислотность уменьшается. 4) Сила к-ты определяется стабильностью образующегося аниона. Чем стабильнее анион, тем сильнее к-та. Стабильность аниона зависит от делокализации (распределения) «-» заряда по всей частице (аниону). Чем больше делокализован «-» заряд, тем стабильнее анион и сильнее к-та. Делокализация заряда зависит: a) от электроотрицательности (ЭО) гетероатома. Чем больше ЭО гетероатома, тем сильнее соответствующая к-та. Например: R – ОН и R – NН 2 Спирты более сильные к-ты, чем амины, т.к. ЭО (О) > ЭО (N). б) от поляризуемости гетероатома. Чем больше поляризуемость гетероатома, тем сильнее соответствующая к-та. Например: R – SН и R – ОН Тиолы более сильные к-ты, чем спирты, т.к. атом S более поляризован, чем атом О. в) от характера заместителя R (длины его, наличие сопряженной системы, делокализации электронной плотности). Например: СН 3 – ОН, СН 3 – СН 2 – ОН, СН 3 – СН 2 – СН 2 – ОН Кислотность <, т.к. увеличивается длина радикала СН 3 – ОН, С 6 Н 5 – ОН, Сила к-ты увеличивается Фенолы являются более сильными к-тами, чем спирты за счет р, p-сопряжения (+М) группы –ОН. Кроме того карбоксилат-анион более стабилен, чем анион спирта за счет р,p-сопряжения в карбоксильной группе. Например: р-Нитрофенол более сильная к-та, чем р-аминофенол, т.к. группа –NО 2 является ЭА. СН 3 –СООН ССl 3 –СООН рК 4,7 рК 0,65 Трихлоруксусная к-та во много раз сильнее СН 3 СООН за счет – I атомов Сl как ЭА. Муравьиная к-та Н–СООН сильнее СН 3 СООН за счет +I группы СН 3 – уксусной к-ты. д) от характера растворителя. Если растворитель является хорошим акцептором протонов Н + , то сила Основания Бренстеда
5) Они делятся на: а) p-основания (соединения с кратными связями); б) n-основания (аммониевые, содержащие атом , оксониевые, содержащие атом , сульфониевые, содержащие атом ) Сила основания определяется стабильностью образующегося катиона. Чем стабильнее катион, тем сильнее основание. Другими словами, сила основания тем больше, чем менее прочная связь с гетероатомом (О, S, N), имеющим свободную электронную пару, атакуемую Н + . Стабильность катиона зависит от тех же факторов, что и стабильность аниона, но с обратным действием. Все факторы, усиливающие кислотность, уменьшают основность. Самыми сильными основаниями являются амины, т.к. атом азота имеет меньшую ЭО по сравнению с О. При этом вторичные амины более сильные основания, чем первичные, третичные амины слабее вторичных за счет стерического фактора, затрудняющего доступ протона к N. Ароматические амины более слабые основания, чем алифатические, что объясняется +М группы –NН 2 . Электронная пара азота, участвуя в сопряжении, становится малоподвижной. Стабильность сопряженной системы затрудняет присоединение Н + . В мочевине NН 2 –СО– NН 2 присутствует ЭА группа > С = О, которая значительно снижает оснóвные св-ва и мочевина образует соли только с одним эквивалентом к-ты. Т.о., чем сильнее к-та, тем слабее образуемое ею основание и наоборот. Спирты
Это производные УВ, у которых один или несколько атомов Н замещены на –ОН группу. Классификация:
I. По количеству групп ОН различают одноатомные, двухатомные и многоатомные спирты: СН 3 -СН 2 -ОН Этанол Этиленгликоль Глицерин II. По характеру R различают: 1) предельные, 2) непредельные, 2) СН 2 = СН-СН 2 -ОН Аллиловый спирт ретинол (витамин А) и холестерин витаминоподобное в-во III. По положению гр. –ОН различают первичные, вторичные и третичные спирты. IV. По количеству атомов С различают низкомолекулярные и высокомолекулярные. СН 3 –(СН 2) 14 –СН 2 –ОН (С 16 Н 33 ОН) СН 3 –(СН 2) 29 –СН 2 ОН (С 31 Н 63 ОН) Цетиловый спирт Мирициловый спирт Цетилпальмитат – основа спермацета, мирицилпальмитат содержится в пчелином воске. Номенклатура: Тривиальная, рациональная, МН (корень+окончание «ол»+арабская цифра). Изомерия:
цепи, положения гр. –ОН, оптическая. Строение молекулы спирта
СН-кислотный Nu центр Электрофильный Центр Кислотный центр основности центр Р-ции окисления 1) Спирты – слабые кислоты. 2) Спирты – слабые основания. Присоединяют Н + лишь от сильных кислот, но они более сильные Nu. 3) –I эффект гр. –ОН увеличивает подвижность Н у соседнего углеродного атома. Углерод приобретает d+ (электрофильный центр, S Е) и становится центром нуклеофильной атаки (Nu). Связь С–О рвется более легко, чем Н–О, поэтому характерными для спиртов явл-ся р-ции S N . Они, как правило, идут в кислой среде, т.к. протонирование атома кислорода увеличивает d+ атома углерода и облегчает разрыв связи. К этому типу относятся р-ции образования эфиров, галогенопроизводных. 4) Смещение элекронной плотности от Н в радикале приводит к появлению СН-кислотного центра. В этом случае идут р-ции окисления и элиминирования (Е). Физические св-ва
Низшие спирты (С 1 –С 12) – жидкости, высшие – твердые вещ-ва. Многие св-ва спиртов объясняются образованием Н-связи: Химические св-ва
I. Кислотно-оснóвные Cпирты – слабые амфотерные соединения. 2R–ОН + 2Nа ® 2R–ОNа + Н 2 Алкоголят Алкоголяты легко гидролизуются, что показывает – спирты более слабые кислоты, чем вода: R– ОNа + НОН ® R–ОН + NаОН Оснóвный центр в спиртах – гетероатом О: Если р-ция идет с галогеноводородами, то присоединяться будет галогенид-ион: СН 3 -СН 2 + + Сl - ® СН 3 -СН 2 Сl НС1 RОН R-СООН NН 3 С 6 Н 5 ОNа С1 - R-О - R-СОО - NН 2 - С 6 Н 5 О - Анионы в таких р-циях выступают в качестве нуклеофилов (Nu) за счет «-» заряда или неподеленной электронной пары. Анионы являются более сильными основаниями и нуклеофильными реагентами, чем сами спирты. Поэтому на практике для получения простых и сложных эфиров используются –алкоголяты, а не сами спирты. Если нуклеофилом является другая молекула спирта, то она присоединяется к карбокатиону: Это р-ция алкилирования (введение алкила R в молекулу). Заместить –ОН гр. на галоген можно при действии РСl 3 , РСl 5 и SОСl 2 . По такому механизму легче реагируют третичные спирты. Р-цией S Е по отношению к молекуле спирта является р-ция образования сложных эфиров с органическими и минеральными к-тами: R – О Н + Н О – R – О – + Н 2 О Сложный эфир Это р-ция ацилирования – введение ацила в молекулу. При избытке Н 2 SО 4 и более высокой температуре, чем в случае р-ции образования простых эфиров, идет регенерация катализатора и образуется алкен: СН 3 -СН 2 + + НSО 4 - ® СН 2 = СН 2 + Н 2 SО 4 Легче идет р-ция Е для третичных спиртов, труднее для вторичных и первичных, т.к. в последних случаях образуется менее стабильные катионы. В данных р-циях выполняется правило А. Зайцева: «При дегидратации спиртов атом Н отщепляется от соседнего атома С с меньшим содержанием атомов Н». Бутанол-2 В организме гр. –ОН превращается в легкоуходящую путем образования эфиров с Н 3 РО 4: IV. Р-ции окисления 1) Первичные и вторичные спирты окисляются СuО, растворами КМnО 4 , К 2 Сr 2 О 7 при нагревании с образованием соответствующих карбонилсодержащих соединений: Нитроглицерин – бесцветная маслянистая жидкость. В виде разбавленных спиртовых растворов (1%) применяется при стенокардии, т.к. оказывает сосудорасширяющее действие. Нитроглицерин – сильное взрывчатое вещество, способное взрываться от удара или при нагревании. При этом в малом объеме, который занимает жидкое вещество, мгновенно образуется очень большой объем газов, что и вызывает сильную взрывную волну. Нитроглицерин входит в состав динамита, пороха. Представители пентитов и гекситов – ксилит и сорбит – соответственно, пяти- и шестиатомные спирты с открытой цепью. Накопление –ОН групп ведет к появлению сладкого вкуса. Ксилит и сорбит – заменители сахара для больных диабетом. Глицерофосфаты – структурные фрагменты фосфолипидов, применяются как общеукрепляющее средство. Бензиловый спирт
Изомеры положения








































Основными направлениями химической термодинамики являются:
Классическая химическая термодинамика, изучающая термодинамическое равновесие вообще.
Термохимия, изучающая тепловые эффекты, сопровождающие химические реакции.
Теория растворов, моделирующую термодинамические свойства вещества исходя из представлений о молекулярном строении и данных о межмолекулярном взаимодействии.
Химическая термодинамика тесно соприкасается с такими разделами химии, как аналитическая химия; электрохимия; коллоидная химия; адсорбция и хроматография.
Развитие химической термодинамики шло одновременно двумя путями: термохимическим и термодинамическим.
Возникновением термохимии как самостоятельной науки следует считать открытие Германом Ивановичем Гессом, профессором Петербургского университета, взаимосвязи между тепловыми эффектами химических реакций ---законы Гесса.
1) по возможности тепло- и массообмена: изолированные, закрытые, открытые. Изолированная система не обменивается с окружающей средой ни веществом, ни энергией. Закрытая система обменивается с окружающей средой энергией, но не обменивается веществом. Открытая система обменивается с окружающей средой и веществом и энергией. Понятие изолированной системы используется в физической химии как теоретическое.
2) по внутренней структуре и свойствам: гомогенные и гетерогенные. Гомогенной называется система, внутри которой нет поверхностей, делящих систему на части, различные по свойствам или химическому составу. Примерами гомогенных систем являются водные растворы кислот, оснований, солей; смеси газов; индивидуальные чистые вещества. Гетерогенные системы содержат внутри себя естественные поверхности. Примерами гетерогенных систем являются системы, состоящие из различных по агрегатному состоянию веществ: металл и кислота, газ и твёрдое вещество, две нерастворимые друг в друге жидкости.
Фаза
– это гомогенная часть гетерогенной системы, имеющая одинаковый состав, физические и химические свойства, отделённая от других частей системы поверхностью, при переходе через которую свойства системы меняются скачком. Фазы бывают твёрдые, жидкие и газообразные. Гомогенная система всегда состоит из одной фазы, гетерогенная – из нескольких. По числу фаз системы классифицируются на однофазные, двухфазные, трёхфазные и т.д.




















































–СН 2 –. Например, гомологический ряд предельных углеводородов:

Например: Винная к-та

D – (+) – ряд L – (–) – ряд
Мезовинная к-та

 С 6 Н 6
С 6 Н 6![]() , X = Наl -, НО -, НS -, NН 2 - и др.
, X = Наl -, НО -, НS -, NН 2 - и др.

Заместители
Ориентанты в С 6 Н 5 -R
I
М
Аlk (R-): СН 3 -, С 2 Н 5 -...
Ориентанты
I рода:
направляют ЭД
заместители
в орто- и пара-
положения
+
– Н 2 , –NНR, –NR 2
–
+
– Н, – Н, – R
–
+
–Н L
–
+

 При одинаковом кислотном центре сила спиртов, фенолов и карбоновых к-т не одинакова. Например,
При одинаковом кислотном центре сила спиртов, фенолов и карбоновых к-т не одинакова. Например, Связь О–Н более поляризуется в фенолах. Фенолы могут взаимодействовать даже с солями (FеС1 3) – качественная реакция на фенолы. Карбоновые
Связь О–Н более поляризуется в фенолах. Фенолы могут взаимодействовать даже с солями (FеС1 3) – качественная реакция на фенолы. Карбоновые
к-ты по сравнению со спиртами, содержащими одинаковый R, являются более сильными к-тами, т.к. связь О–Н значительно поляризована за счет –М эффекта группы > С = О: г) от введения заместителей в радикал. ЭА заместители увеличивают кислотность, ЭД заместители уменьшают кислотность.
г) от введения заместителей в радикал. ЭА заместители увеличивают кислотность, ЭД заместители уменьшают кислотность.
к-ты увеличивается и наоборот.


3) циклические, 4) ароматические. 3) К непредельным циклическим спиртам относятся:
3) К непредельным циклическим спиртам относятся:
 Инозит
Инозит![]()
СН 3 -СН 2 + + ® СН 3 -СН 2 + - - Н СН 3 -СН 2 -О-R
Простой эфир
 СН 3 -СН = СН -СН 3
СН 3 -СН = СН -СН 3 3)